
作者:Jerry发呆
有时候跟非药学专业的同学聊天,他们会问我做新药难不难。我可以罗列一堆临床试验成功率数据来论证做新药有多艰难,不过拿PARP抑制剂的研发过程来举个例子更合适不过了。
PARP的全称是多聚ADP核糖多聚酶,上世纪60年代就被发现,但直到80年代初期通过化学探针抑制酶活性才确定了该酶的功能。科学家们早在几十年前就开始尝试研发靶向PARP酶家族的药物,之后对PARP抑制剂主要作用机制的理解则不断深入。
科研人员最初认为PAPP抑制剂的药效是单纯的通过抑制PARP催化活性产生的,使DNA单链损伤无法得到修复,而DNA复制时产生的复制叉行进至PARP结合的损伤位点时,可能会引起复制叉被破坏进而引起DNA双链损伤,从而引发DNA损伤反应,诱导细胞凋亡 [1]。
但最近几年的多项研究表明,一些PARP抑制剂能够使PARP1无法从DNA损伤位点脱离,影响PARP1酶的催化循环过程,进而影响DNA稳定性并导致细胞毒性作用 [2-4]。这种作用机制与DNA拓扑异构酶II的作用机制比较类似,该酶抑制剂的作用机制同样是使DNA修复蛋白无法从双链DNA上脱离。
依据PARP抑制剂的药理机制,科学家们提出了PARP抑制剂在肿瘤领域的两种临床应用策略。
第一种策略是PARP抑制剂与化疗药物联用治疗各种类型的癌症,该策略如果成功毫无疑问将会带来巨额的商业利润,因此制药公司开始投入巨额经费研究PARP抑制剂与各种化疗药物的联用。但科研人员在研究过程中发现化疗与PAPP抑制剂联用的毒性太大,两类药物产生的协同作用可以产生严重的骨髓抑制,很难找到PAPR抑制剂与化疗药物联用的安全药物剂量。
另一种策略是基于合成致死(synthetic lethality)的理念。Synthetic lethality经常被翻译为合成致死,个人认为翻译为协同致死或者综合致死可能更为贴切,但出于阅读习惯考虑,本文沿用合成致死。这个策略的出发点是:单个基因突变不会对细胞生存产生明显影响,但两种关键基因损伤同时发生会导致细胞死亡。
肿瘤细胞具有两种DNA损伤修复能力,一种是修复双链DNA断裂的同源重组 (HR) 修复,另一种是PARP参与的单链DNA修复。如果同时削弱肿瘤细胞的两种DNA损伤修复机制可以诱导肿瘤细胞凋亡,这正是合成致死理念的最好诠释。
这也意味着如果肿瘤细胞HR修复功能本身就存在问题,那么PARP抑制剂就可以作为单药使用。而在此之前科学家们就发现存在BRCA突变的肿瘤细胞HR修复功能会存在缺陷。2005年Nature同期发表了两篇文章证实了存在BRCA突变的肿瘤细胞对PARP抑制剂非常敏感 [5-6]。该研究也引发了PARP抑制剂研究的第二轮热潮。

by Jerry
与联合化疗的方案相同,合成致死策略的早期临床研究并没有获得积极的疗效数据,导致PARP抑制剂的两种临床应用之路在当时看来似乎都行不通。不过进行临床前动物模型研究的科研人员也发现,PARP抑制剂与化疗联用和单药使用时的作用模式存在明显差别。小鼠实验表明,如果PARP抑制剂联用某种DNA损伤药物,PARP抑制剂的给药剂量必须非常低,而且用药周期必须很短,否则会引起实验动物的死亡。而如果单独使用PARP抑制剂的话,给药剂量必须非常高,而且必须保证足够长的用药周期才能抑制肿瘤的生长。
换句话说,第二种策略是单独用药,安全性不存在问题,理论上可行性更高。只不过科研人员最初在单独使用PARP抑制剂时,给药剂量是参照与化疗联用时使用的剂量,认为低剂量及短期给药就足以发挥药效。由于当时人们并不知道如何合理使用该类抑制剂,由此导致临床试验设计上存在缺陷,试验失败也是情理之中的事情。
I、波 折
PARP抑制剂的研发过程真可谓是一波三折,虽然对于用药剂量问题的研究已经比较深入,但接下来的临床开发依然十分不顺利。该类药物开发过程中最重大的挫折来源于BiPar Sciences开发的抑制剂BSI-201。2009年4月赛诺菲以大约5亿美金的价格收购BiPar Sciences获得了该药的权益。赛诺菲当时以为自己捡了个大便宜,很快在ASCO2009大会上公布了该药 (后更名为iniparib) 的II期临床试验数据,称iniparib联合吉西他滨和卡铂能够延长三阴乳腺癌患者的生存期,而且PARP抑制剂的联用方案并没有产生明显安全性问题。2010年,赛诺菲又在ESMO会议上公布了该试验的具体数据,称该联用方案可以使患者的生存期延长近5个月。
但接下来发生的事情却让赛诺菲高兴不起来了。2011年初,赛诺菲宣布iniparib在一项III期试验中无法延长三阴乳腺癌患者的生存期 。由于iniparib之前的影响力太大,该结果的公布瞬间浇灭了整个制药行业对PARP抑制剂的开发热情。
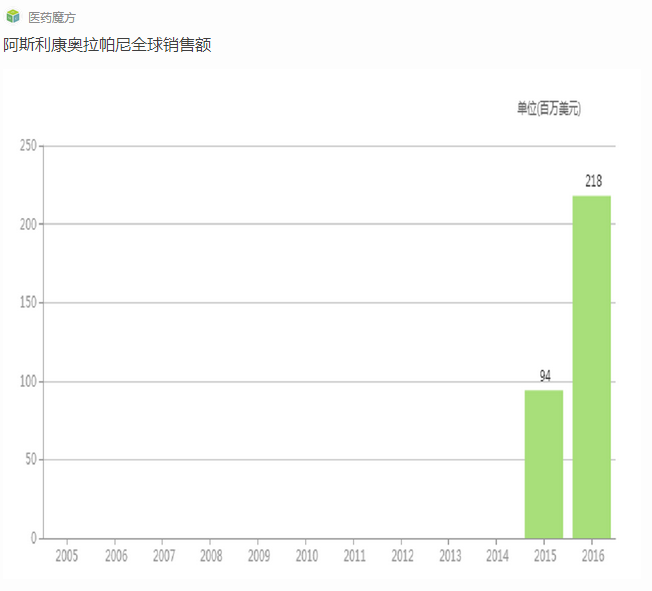
阿斯利康在同一时间宣布停止自己PARP抑制剂olaparib用于BRCA阳性乳腺癌患者的开发。而且在2011年末,阿斯利康宣布olaparib一项针对卵巢癌患者的II期临床试验失败,选择停止该适应症的开发。数据显示虽然olaparib能够延缓卵巢癌患者疾病进展,但无法延长患者生存期。
在同一时间,辉瑞将自家的rucaparib卖给了Clovis Oncology,而默克也在2012年将niraparib卖给了Tesaro。由于存在太多的不确定性,大多数制药公司不太愿意再启动新的临床研究。
然而,2012年拉瓦尔大学医学中心的Kaufmann发表文章称赛诺菲的iniparib并不是真正的PARP抑制剂 [7]。可以说这一发现让肿瘤学家和投资人倍感欣慰,因为很早之前他们就觉得iniparib的临床数据非常奇怪,与其他多个PARP抑制剂的动物实验数据并不一致,而且临床研究中iniparib联用卡铂和吉西他滨并不会产生明显的骨髓抑制现象。但由于赛诺菲和BiPar当时没有公开临床研究过程中的的用药剂量等关键数据,因此科学家们也没有确认iniparib的体外活性。
其实Clovis Oncology在购买辉瑞的rucaparib权益之前了解到iniparib不会产生骨髓抑制作用,他们就开始怀疑iniparib不是真正的PARP抑制剂,这也可能是Clovis Oncology果断从辉瑞购买rucaparib继续开发的原因。
2012年末,阿斯利康在对olaparib卵巢癌II期临床数据进一步分析之后发现,部分患者使用olaparib后确实能够产生明显获益,随即在ASCO2013大会上公布了回顾性分析结果——携带BRCA突变的卵巢癌患者相比不携带突变患者对olaparib的响应率更高,而且相比安慰剂组,携带突变的患者能够使PFS(无进展生存期)延长6.9个月。更重要的是,携带BRCA突变并使用Olaparib的患者相比安慰剂组可使生存期延长3个月。受以上数据的鼓舞,阿斯利康2013年下半年启动了两项III期研究以评价olaparib单药维持治疗携带BRCA突变的卵巢癌患者的疗效。
2014年12月21日,FDA批准olaparib上市用于既往经至少3次化疗治疗失败的BRCA胚系突变晚期卵巢癌患者的治疗。2016年12月19日,FDA批准Clovis的PARP抑制剂rucaparib上市,用于治疗已接受两种及以上化疗方案的且存在胚系或者体细胞BRCA基因突变的晚期卵巢癌患者的治疗。
尽管早期的临床研究主要集中于对存在BRCA胚系突变的肿瘤的有效性研究上,但后来发现不携带该类突变的患者也能够产生应答 [9]。其实从PARP抑制剂和合成致死的作用机制的角度很容易解释这一现象。
PARP抑制剂可以抑制PAPR的催化循环过程,使PARP无法从已结合的DNA上脱离,由此引发DNA损伤反应 (DDR),而BRCA1/2 参与的同源重组修复 (HRR)是DDR中最佳的DNA修复手段,能够有效修复和重启由于PARP抑制剂引发的复制叉行进中止。如果HRR机制存在缺陷的话将有可能产生合成致死效应,诱导肿瘤细胞凋亡。然而BRCA1/2 并不是HRR机制中唯一的重要组分,参与编码同源重组修复通路中的其他重要蛋白的基因突变也有可能引发HRR机制缺陷,产生合成致死。因此理论上来讲BRCA1/2并不是PARP抑制剂的唯一生物标志物 (biomarker)。
最终,临床试验也证实了PARP抑制剂的有效性并不局限于BRCA基因突变。因此FDA在今年3月27日批准Tesaro公司的niraparib上市,用于接收铂类药物治疗后完全应答或部分应答但又疾病复发的成人卵巢上皮癌、输卵管癌和原发性腹膜癌患者的维持治疗。8月17日,olaparib与niraparib相同的适应症获得FDA批准。
其实PAPR抑制剂有效性超越BRCA基因突变局限性这一理念的验证,不仅扩大了PARP抑制剂的应用范围,也同时带动了PARP抑制剂生物标志物的研究以及恢复了科研人员对整个合成致死领域研究的信心。
II、合成致死的复兴
当前主流的肿瘤靶向药物的开发主要是基于原癌基因成瘾性 (oncogene addiction)的机制来研发的,也就是说通过抑制肿瘤生长依赖的原癌基因或者相关通路来研发药物。尽管这些药物对某些类型的肿瘤有效,但并不是所有肿瘤都携带具有成药性的功能获得性原癌基因突变,这些肿瘤中携带的非原癌基因突变可能与其他肿瘤特异性基因突变相互作用,两类基因同时抑制时会诱导细胞凋亡,产生合成致死效应,因此合成致死理念的应用可大范围的扩展抗肿瘤药物研发的潜在靶标。
但由于PARP抑制剂开发过程中的波折,尤其是iniparib乌龙事件的影响,即使在olaparib获批上市之后,制药行业对于基于合成致死理念的新药研发热情依然不高。除了iniparib的影响,该领域研究进展比较缓慢的一个非常重要的原因是缺乏高效的研究工具。
直到最近几年,制药公司和学术机构的研究人员一直在使用RNAi技术来寻找抗肿瘤药物靶点,通过大批量筛选能够抑制基因表达的siRNA/短发夹RNA干扰文库来筛选能够产生合成致死作用的基因。但RNAi技术存在一个致命的缺陷:脱靶效应太强。基于RNAi技术的实验能够产生大量的假阳性结果,因此使靶点的验证变得非常困难。这也是很多专注于合成致死领域研究的生物制药公司始终没能取得很大进展的主要原因之一。
但最近一两年内出现了另一批专注于合成致死领域的生物制药公司,这些公司使用的是比RNAi高效的多的研究工具: CRISPR。自从2013年CRISPR基因编辑工具诞生以来,该技术对基础科研,农业,医药等领域产生了极大的影响。笔者在之前的文章里非常详细地介绍了该技术的基础概念,发展过程以及对多个领域的影响。通过CRISPR gRNA分子库来寻找能够产生合成致死相互作用基因也成了该类公司融资的基础。
今年6月Repare首轮融资6800万美金,这应该是2017年上半年生物制药行业金额最大的一笔早期融资。上个月KSQ Therapeutics融资7600万美金,部分资金将用于合成致死药物靶点的研究。
学术机构和制药公司的科研人员通常使用两种基于CRISPR技术的策略来筛选合成致死药物靶点。第一种是使用CRISPR gRNA分子库对细胞关键基因逐个进行敲除,并与未进行基因敲除的细胞进行对比,寻找能够影响细胞功能的基因。而另一种策略是使用的是包含或者不包含已知的合成致死相关基因突变 (比如BRCA基因突变) 的两种细胞株,使用CRISPR分子库逐个敲除两种细胞株的基因,通过对比两类细胞基因敲除之后的影响来筛选靶点。
以上两种方法相比RNAi技术都能获得更优质的药物靶点,但在技术的具体应用过程中每个公司都各有不同。例如Tango公司首先会寻找一部分病人,并使用与每位病人基因相匹配的细胞株进行实验,利用经过成药性优化的CRISPR gRNA分子库 (只筛选可能成药的基因靶标) 来获得药物靶点。而KSQ则是对将近600多个细胞株的全基因组基因逐个进行敲除 (人大约有19000个基因,这种敲除效率在CRISPR出现之前是无法想象的),从而获得大量的基因敲除对细胞功能影响的数据。
尽管CRISPR技术极其高效,但并非完美无缺,其中最重要的一点是基因敲除与使用RNAi进行基因沉默以及通过小分子抑制蛋白功能产生的效应可能完全不同,因此如何对试验结果进行解读变得十分重要。而且合成致死相互作用的筛选也存在诸多问题,比如很多合成致死相互作用是条件依赖性的,在细胞环境改变之后其相互作用可能消失。
无论是CRISPR技术还是RNAi,都是通过细胞水平的功能研究来获得药物靶点,细胞水平的功能影响并不一定意味着能够对整体动物或者人体中的肿瘤产生影响,即使获得了大量的潜在药物靶点,也必须经过繁琐的进一步靶点验证过程,基于靶点的药物设计合成研究过程,以及临床试验研究。寻找药物靶点只是万里长征的第一步。
近两年合成致死领域的生物技术公司融资情况

III、未 来
近几年,肿瘤免疫疗法的迅速发展已经对抗肿瘤领域的新药研发产生了极大影响。几乎每一个涉足抗肿瘤新药领域的制药公司都不得不去考虑现有疗法如何与已上市的肿瘤免疫疗法联用。
存在BRCA1/2突变以及HRR缺陷的肿瘤通常具有更高的突变率,因此可能产生更多的新抗原 (neo-antigen),从而引发更强烈的免疫反应。所以理论上来讲,PARP抑制剂或DNA损伤抑制剂联用PD-1或者CTLA-4抑制剂治疗存在HRR缺陷的肿瘤能够提高药物疗效/患者响应率 [9-10]。而且有研究显示BRCA缺陷能够引发STING依赖性的固有免疫应答,这也为PARPi/肿瘤免疫疗法提供了依据 [11]。
尽管PARP抑制剂/肿瘤免疫疗法联用理论上可行,而且已有制药公司开始进行该方面的临床研究(例如NCT02734004,临床1/2期,olaparib/durvalumab),但仍然需要经历漫长的临床研究才能确定该策略的有效性与安全性。早期PARP抑制剂/化疗联用临床研究的失败(化疗/PARP抑制剂联用从理论机制上讲更加合理)也提醒我们不应该过度的乐观 [12]。
国内做PARP抑制剂的制药公司以及学术机构也非常多,进入临床的包括百济神州的BGB-290,恒瑞的SHR-3162等等,需要指出的是不同结构的PARP抑制剂疗效和作用机制差别可能会特别大。
国内注册申报的PARP抑制剂
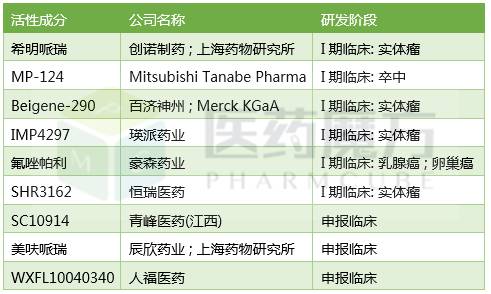
来源:医药魔方发现
PARP抑制剂不仅能够抑制该蛋白的活性,更重要的是能够抑制PAPR从DNA上脱离,而不同的抑制剂这两方面性质可能存在明显差异。例如临床研究中使用替莫唑胺 /veliparib联用,理论上来说PARP抑制剂可以通过抑制PARP的DNA脱离来增强DNA烷化剂替莫唑胺的DNA损伤作用,但是由于veliparib抑制PAPR蛋白DNA脱离的作用非常弱,所以导致veliparib无法提高替莫唑胺的疗效 [13]。
我的同学当中也有人在做PARP抑制剂,其实从某种层面上来讲做药化方面的研究并不是一件容易的事情,有些化合物的合成过程本身就让人非常头疼,经过一两年的折腾合成出几十个化合物之后,几轮筛选和结构优化也难找到活性高选择性好的小分子。即使得到活性比较好的化合物,后续的细胞水平实验,动物模型安全性有效性实验的过程中也存在重重障碍,ADMET性质的优化也会很让人崩溃。
对于PARP抑制剂来说临床研究则更加困难,从化疗联用以及2005年Nature文章[5-6] 验证的PARP抑制剂/BRCA合成致死理念到olaparib的上市总共花费了超过10年的时间。10年之后我们才逐渐理解临床中如何合理的应用PARP抑制剂。我们很难预计该领域未来还会遇到多少阻碍,但借用我导师的一句口头禅——Always hope for the best.
参考文献
1 Nature. 434, 917–921. 2005.
2 Cancer Research. 72, 5588–5599. 2012.
3 Molecular. Cancer Therapy. 13, 433–443. 2014.
4 Science. Translational. MedicinAe. 8, 362ps17. 2016.
5 Nature. 434: 913–917. 2005.
6 Nature. 434: 917–921. 2005.
7 Clinical Cancer Research. 18(6):1655-62. 2012.
8 Lancet Oncology. 12, 852–861. 2011.
9 Cancer Immunology Research. 3:1257–1268. 2015.
10 Biochem. Biophys. Res. Commun. 463, 551–556. 2015.
11 Journal of the National Cancer Institute. 109:djw199. 2016.
12 Crit. Rev. Oncol. Hematol. 108: 73–85. 2016.
13 Future Oncol. 13: 307–320. 2017.Q
