
根据国家癌症中心最新发布的《2017中国肿瘤现状和趋势》,结、直肠癌发病率在中国男性患者中位居第五,在女性患者中则进入前三名。以总体肠癌发病人数而言,中国患者人数已跃居世界第一位。90%的恶性结肠肿瘤均由结肠息肉发展而来,其中腺瘤性息肉的恶变几率远远超过普通炎性息肉。临床研究表明结肠癌发病常常表现出家族性,且60%患者体内存在腺瘤性息肉易感蛋白(Adenomatous Polyposis Coli,APC )编码基因突变,该突变在癌细胞转移过程中扮演了重要角色。
通过大量结肠息肉、癌症患者的活检病理分析,以及长达七年的实验数据验证,上海交大医学院张健教授首次以APC-Asef蛋白相互作用为靶标,设计首创全新抑制剂,为降低癌细胞转移和提升生存期限带来了新的曙光。张健教授多年来一直从事重大疾病的精准靶标识别和“first-in-class”药物发现研究;基于多年的临床一线大数据分析,张教授还创立了有别于传统靶蛋白抑制剂/激活剂的变构药物设计体系,形成了完整的变构药物开发方法论,包括靶蛋白变构位点识别方法、筛选方法、功能评价标准,为攻克肿瘤等重大疾病带来了新的希望。

▲张健教授2007年获得上海药物所博士学位后,前往美国密歇根大学肿瘤中心进行博士后研究,其团队发展的系列变构药物设计方法得到广泛应用。张教授已入选国家自然科学基金优秀青年基金、教育部青年长江学者和中组部万人计划青年拔尖人才,2017年获得药明康德生命化学研究奖。
发现神奇靶标,七千人基因组中披沙拣金
Q:请您介绍一下团队研发的新药,通过何种机制抑制结肠癌细胞转移?
张健教授:APC是一个具有多个结构域的大型蛋白,在结肠癌发生、发展中具有多种功能。正常的APC蛋白并不引起结肠癌转移,然而当结肠癌患者体内的APC蛋白因突变而成为异常的截短体,则会异常结合并激活下游Asef蛋白引发结肠癌转移。Asef蛋白(APC-stimulated guanine nucleotide exchange factor)属于鸟苷酸交换因子家族,正常的Asef蛋白处于自我抑制状态。但是与被截短的APC蛋白结合后,Asef蛋白会被持续激活其鸟苷酸活化因子活性,并促进癌细胞转移。
我们解析了APC-Asef蛋白复合物晶体结构,发现Asef蛋白通过ABR结构域结合在APC蛋白的ARM结构域。我们发展了诱导契合的蛋白相互作用界面小分子设计方法,将APC原本难以结合小分子的宽大平坦口袋,转变为环形类药性口袋。由此获得的低毒、高效创新抑制剂MAIT-203,可特异性阻断APC-Asef蛋白相互作用,阻断结肠癌的恶性转移,且不影响APC和其他蛋白的亲和力。我们还通过该化合物识别了APC-Asef蛋白相互作用介导的潜在下游通路蛋白。

▲APC蛋白ARM结构域与Asef蛋白ABR结构域结合
(图片来源:《Cell Research》杂志)
Q:发现APC-Asef相互作用这样一个靶标,您的主要灵感来自于何处?
张健教授:相对于参考科学文献,我们团队更热衷于从一线临床数据中寻找靶标。每位癌症患者的基因组特点不尽相同,他人验证的靶标并不一定适用于每个患者,还要考虑种族差异和动物模型的差异。所以识别各种肿瘤亚型所特有的全新靶标,已经成为靶向药物开发的重要前提。我们利用交大医学院众多附属医院临床资源丰富的优势,通过与临床科室合作并进行大规模病人样本筛查,解析了33类肿瘤的7000例临床病人基因组。
我们发现肿瘤基因变化主要富集在蛋白的活性底物位点及变构位点。我们设计了一套识别不同肿瘤敏感靶标的方法AlloDriver,主要参照基因变化在变构位点的映射分布。利用此法,我们发现已知治疗精神类疾病的药物靶点PDE10A,同是也是非小细胞肺癌的全新靶标,并通过化学生物学方法证明:靶向PDE10A已上市药物可有效杀伤非小细胞肺癌细胞。
另外相较于改进既有靶标和药物,我们团队更偏好于寻找全新靶标和“first-in-class”先导化合物。相对于“试对“,我们更喜欢“试错”,所以我们希望完成临床前的靶标识别和药物发现迭代的源头创新工作,再由更适合药物开发的企业承担制剂合成和临床推进的角色。如今医药监管机构推出许多鼓励药物创新的政策,所以我们也希望在这个利好的大环境下,不断开发更多为患者造福的原研新药。

▲张健教授团队发表的论文,绘制了多种蛋白激酶的变构位点。(图片来源:《Nucleic Acids Research》)
变构药物:发掘蛋白活性位点之外的蹊径
Q:除了单纯发现小分子化合物,您的团队还很善于总结全新的方法论。变构药物设计是您团队的一大成就,请问这类有别于传统激活剂/抑制剂的变构小分子有哪些特点?
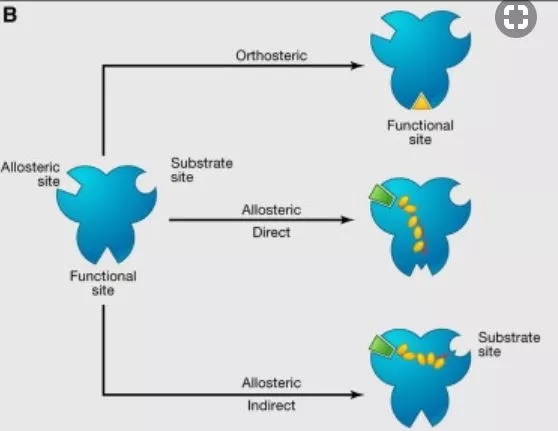
张健教授:对于癌症等重大疾病的靶向疗法,靶标通常是靶蛋白的正位活性位点,而这些活性位点在人体内不同蛋白质中可能呈现同源性高、进化保守的特点。而与此相对的,则是人体内同一家族、或者同一蛋白的不同亚型之间,变构位点却呈现出高度多样性。因此针对变构位点设计的变构药物,具有高度特异性且脱靶毒性风险下降。
另外针对靶蛋白活性位点开发的靶向药物,多为竞争性抑制剂/激活剂,所以患者需要服用远超过内源性配体的药量,才能达到效果。考虑到不少癌细胞表面的靶蛋白数量可能远超正常细胞,以及药物递送过程中的损失,伴随患者服药的毒副作用风险进一步提升。然而变构药物并非竞争性调节剂,它们发挥药效的方式,是改变靶蛋白与内源性配体的结合效率,或者直接改变靶蛋白构象及其信号传导通路。因此变构药物服药剂量可大大降低,减少人体代谢压力。
▲蛋白质正位活性位点 & 变构位点(图片来源:《Cell》杂志)
另外针对靶蛋白活性位点开发的竞争类药物,只能模仿靶蛋白结合内源性底物的通路,具有抑制剂或激活剂的单一功能;而针对非竞争类变构位点开发的调节剂,却可以有抑制/激活/中性调节三种方向,无疑大大拓展了靶向药物的开发空间。如果未来变构类小分子药物再与抗体类生物药发生偶联,则会对癌症等重大疾病的靶向疗法产生重大变革。
Q:请您谈谈变构药物研发的挑战,以及您团队由此总结出的一套方法论。
张健教授:变构药物设计实际上是一个系统工程,自1901年变构现象发现以来一直存在着多个亟待解决的科学问题,例如变构位点难以发现,变构位点的小分子药物筛选成功率低,结构优化效果差等。针对变构药物开发中的这些关键问题,我们系统发展了一套完整的变构药物开发方法论,包括变构综合数据库ASD、变构性能评价方法Asbench、变构位点识别方法Allosite、变构小分子集中库构建方法Allolike、变构小分子筛选评价方法Alloscore和变构组学分析方法Allosterome,构建了完整的变构先导化合物合理发现流程。
自2009年我们的变构系列方法发展至今,已有超过100多个国家的50000余名用户收益于我们的技术,对国际变构药物设计及其机制研究领域的发展起到了重要推进作用。我们构建的ASD数据库是国际上首个变构领域数据库,为变构机制研究以及变构药物合理发现提供了丰富系统的数据,一举打破了该领域中曾经存在的瓶颈,Pfizer等20余家跨国制药公司8年来一直是我们的用户。在此基础上建立的Allosite作为国际上首个公开使用的变构位点识别方法,自2013年发布至今,已经在至少12个全新靶蛋白中发现理想位点,包括隐藏在蛋白内部空间中的变构位点。在合理发现变构位点的基础上,我们进一步发展了Allolike和Alloscore,筛选和优化作用于变构位点的先导化合物。利用上述方法,我们团队先后发现了多个变构活性小分子:如CDK2变构抑制剂、STAT3变构抑制剂等。

▲图片来源:Alloscore官网
新方向:代谢性疾病&儿童实体瘤全新变构靶标
Q:除了现有研究成果继续向临床推进,您未来还会重点关注哪些方向?
张健教授:我喜欢不断尝试全新的领域。之后我们关注的方向会包括代谢、衰老相关的疾病。这些领域的科研竞争其实很激烈,常常会看到一些似是而非的数据。经过近八年研究,近期我们已经获得了一个优秀化合物,也是基于变构技术开发的表观遗传激动剂,不仅可用于治疗肝癌、结肠癌,还能够缓解甚至抑制脂肪肝、其他非病毒性肝炎、高血脂相关疾病。目前该成果的学术论文还在数据补充阶段,相关小分子也已经进入临床前试验。
另外我们还希望关注一些罕见肿瘤,比如罕见头颈癌、儿童实体瘤等。目前我们发现不少儿童实体瘤微环境与成年人差异非常大,相关的基因靶标和代谢通路也迥然相异。然而像多数儿童用药一样,儿童肿瘤的治疗方式也多承袭成年人处方,所以我们希望能够在这些领域找到全新的靶标和药物,大大提升儿童癌症的疗效和预后结果。
Q:工作之外您还有哪些兴趣爱好?请谈谈这些爱好对于您科研和生活的帮助。
张健教授:除了科研,我也非常喜欢阅读日本的侦探小说,许多畅销侦探小说家的作品已通关阅尽。西方恐怖小说热衷探讨宗教灵魂相关的主题,但日本侦探小说却喜欢挑战读者的想象力和智商极限,很少有人能够猜到日本悬疑故事的结尾。阅读经历对于科研工作也有不少启发,因为科研也需要挑战极限又丝丝入扣的布局与推理方式。
近来我最喜欢的一个故事,是东野圭吾的《解忧杂货店》。不同于紧张的侦探故事,这本书聚焦了一组又一组超越时空的玄幻对话。回信者不经意间的精神指点,却能够使身处逆境的写信人发生命运的转折。我也会吸取其中充满阳光的思想,借鉴自己对于年轻学生的指导与培养。
孔子云:四十不惑。未来在科研领域我们将不断有突破与挑战,但我觉得悲欢离合的人生际遇,在人到中年之后则波澜渐平。所以我会更加坦然、理性地看待成败得失,而不会将每一个惊喜与失落放大。不论是科研还是生活,都是通往宠辱不惊的修炼。回首向来萧瑟处,也无风雨也无晴。
