
作者: 刘子晨
这是一场触及药品权利持有本质的变革,MAH制度的出台将使得药品核心权益所属在法律层面得以进一步明确,紧随其后而来的,则是未来中国医药生态环境下研发、生产、销售等各领域企业生产关系与市场结构的巨大变化。
2017年10月23日,国家食品药品监督管理总局(下称CFDA)公布了《中华人民共和国药品管理法修正案(草案征求意见稿)》,第一点修正,便是在总则之后增加了第五条规定:“国家实行药品上市许可持有人制度,药品上市许可持有人对药品安全、有效和质量可控承担法律责任。”
这意味着,从2015年11月便开始在全国十省市试点的药品上市许可持有人制度(Marketing Authorization Holder, MAH),终于明确释放出了即将全国推行的信号。
相较于此前药品上市许可与生产许可捆绑的模式而言,MAH制度的出台使得上市许可与生产许可分离,无疑被赋予了更多的期望:鼓励药物研发创新、保障药品供应、遏制低水平重复建设、促进生物医药产业发展。药品研发机构、科研人员以及药品生产企业的研发热情被进一步调动,而诸多CMO/CDMO企业则看到了作为受托方所面临的巨大商业机会。
而更关键的地方在于,这是一场触及药品权利持有本质的变革,MAH制度的出台将使得药品核心权益所属在法律层面得以进一步明确,紧随其后而来的,则是未来中国医药生态环境下研发、生产、销售等各领域企业生产关系与市场结构的巨大变化。
从目前来看,药品上市许可持有人制度的全国推行已是一个必然事件,其对于整个医药行业的促进作用也毫无疑问。但必须要认识到的是,这绝对不是一个终点,法律法规的完善、配套措施的跟进、各方职责的分辨,以及MAH制度所涉及的监管有效性、药品安全、风险分担等社会问题,都尚需要进一步厘清。无论是对于委托企业还是受托企业来说,需要补的课都还有太多。
1
打破“卖青苗”的窘境
打破传统利益藩篱,鼓励新药研发创新。就当下中国医药产业的生态环境而言,这应该是MAH制度出台最根本的原因。
一方面,传统制药企业各自为战,产能的冗余、品质的落后、品种的重复,成为制约中国制药企业往前发展的重要因素;另一方面,创新药企研发成果转换困难,动力不足,创新水平不高,中国想要在制药领域快速赶超,就必须解决这些问题。
而MAH制度最早正式在官方文件中的出现,还要追溯到2015年8月备受行业关注的国发44号文《国务院关于改革药品医疗器械审评审批制度的意见》。44号文明确提出,要开展药品上市许可持有人制度试点,并对此进行了进一步细化解读:允许药品研发机构和科研人员申请注册新药,在转让给企业生产时,只进行生产企业现场工艺核查和产品检验,不再重复进行药品技术审评。
这一点,正是将药品上市许可与生产许可捆绑模式解绑的重要政策基础,亦是解决提高创新水平、促进激进型并解决产能冗余的基础。
在此之前,我国对于国产药品仅允许药品生产企业在取得药品批准文号,并经过药品生产质量管理规范(GMP)认证之后,方可进行生产。这就使得在实践中,药品研发机构和科研人员无法取得药品批准文号,新药研发机构获得新药证书后,只能将相关药品技术以一定价格转让给药品生产企业,业内俗称“卖青苗”。
而对于药品研发机构以及科研人员来说,“卖青苗”无疑是一件极不划算的事情:成果转化率低,回报率小。相对于在前期研发投入的大量技术成本,几百万元的技术转让价格显得颇为低廉,更遑论“卖青苗”所造成的技术成果流失等损失。
“不利于鼓励创新,不利于保障药品供应,不利于抑制低水平重复建设。”在CFDA发布的《药品上市许可持有人制度试点方案》政策解读中,CFDA连用三个“不利于”,一针见血地道出过去药品注册与生产许可捆绑模式的弊端所在。药品上市许可持有人制度的出现,便是CFDA提出的这种弊端的解决之道。
2015年11月4日,第十二届全国人民代表大会常务委员会第十七次会议审议通过《关于授权国务院在部分地方开展药品上市许可持有人制度试点和有关问题的决定》,正式授权国务院在北京、上海、河北、天津等十省市开展MAH制度试点,为期三年,允许药品研发机构和科研人员取得药品批准文号,对药品质量承担相应责任。同时,为MAH制度的试行奠定了法律基础。
而自MAH制度试行以来,其取得的成绩也显而易见。在2017年6月份于上海召开的“药品上市许可持有人制度高峰论坛”中,CFDA药品化妆品注册管理司副司长杨胜透露了一组数据:截至2017年5月31日,根据试点方案申报受理的注册申请共有381例。
值得注意的是,在这381例注册申请中,除了常规的以生产企业为主体的申请仍占多数之外,也已经出现了大量以研发机构为主体的申报案例,共计有142例,占比为37%。除此之外,还出现了一例以科研人员为主体的申报案例,这在之前是绝无仅有的。
与此同时,各试点城市也纷纷加快当地MAH制度的推广进程,一些成果也已经开始显现出来。从发布细则的速度来看,毫无疑问是上海最快,“我们不光发布了实施方案,还同时出台了办事指南、相关政策解读,时间我记得特别清楚,2017年8月3日,我们整理出文件后领导一批我们就马上发布出来了”。上海市食药监局副局长陈尧水此前在“药品上市许可持有人制度高峰论坛”如此表示。
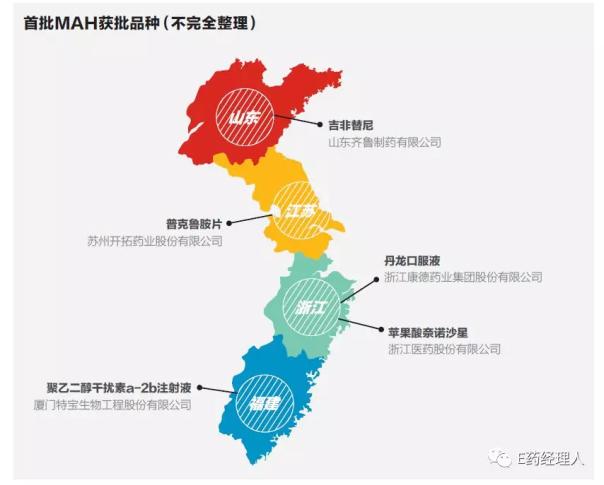
而山东则在行动上占据了先机。2016年12月23日,山东省齐鲁制药研发的抗肿瘤药吉非替尼及片剂经总局批准取得持有人文号,成为国内首个药品上市许可持有人制度试点品种。而随后,2017年3月27日,浙江医药新昌制药厂的苹果酸奈诺沙星原料药及胶囊剂经CFDA批准,成为了全国首个创新药MAH试点品种。同样是浙江省,浙江康德药业申报的丹龙口服液则成为了国内首个中药新药上市许可持有人品种。
2
四方皆赢
理论上来说,这是一场四方皆赢的制度尝试。
首先受益的,自然是此前并不具备药品生产资格的研发企业与科研人员。MAH制度之下,研发企业与科研人员可以作为上市许可持有人持有药品批准文号,再分别委托药品生产企业与药品经营企业进行生产和销售,从而直接获得收益,其新药研发行为的积极性自然也大幅提高。
除此之外,新建生产线所需要的大量费用得以节省下来,则是一众以研发能力为核心的创新药企最看重的因素之一。华领医药此前在进行2型糖尿病用药GKA葡萄糖激酶激活剂HMS5552的二期临床试验时,是否要自建生产基地便是最为困扰华领医药CEO陈力的问题,“在没有完成二期临床之前,要投2个亿去建生产厂是不太可能的,但如果做完二期之后再启动建厂,那肯定要耽误两三年时间。”华领医药并非个例,几乎所有研发型企业都会遇到类似问题,而MAH制度的出现则是柳暗花明又一村:既然有那么多CMO企业可供选择,何必选择自己建厂这一耗时耗力的笨办法呢?
其次则是药品生产企业。尽管看上去与MAH制度关系不大,但实际上,“即便是在生产企业中,也有10%左右是委托生产。”杨胜如此表示。而这与当下诸多医药企业不断战略转型有关,例如一些传统的化药生产企业计划向大分子药、单抗药进军,或是传统的中药企业开始做化学药品,通过委托有生产能力的企业来生产,就无需再新建生产线,从而减免大量成本。
即便是出于未来生产转移等因素考虑,MAH制度对于药品生产企业来说同样至关重要,齐鲁制药所申报的全国首个获批MAH品种吉非替尼便是如此。“吉非替尼为公司持有,由自己生产,但是考虑到该产品未来市场需求有大的增长,可能需要扩产建设新厂区,需要到不同的地方建厂,拥有持有人文号的话,有利于生产转移。”齐鲁制药药品研发负责人在接受E药经理人采访时表示。
而从国家层面来说,药品创新的动力被大幅提升,药品质量将有显著改善,产业集中度将进一步提高,各方面资源进一步得到优化配置,专业分工日益明显,重复投资和建设在很大程度上得以避免。
但受益最为直接和明显的,则还是市场上的CMO/CDMO企业,即医药合同定制研发生产。
“MAH制度试点推出之前,我们多数的业务与订单都来自海外,MAH制度的施行对我们发展国内业务起到了巨大的推动作用。”CDMO企业凯莱英医药集团首席运营官杨蕊在接受E药经理人采访时表示。
这绝对是一块不容小觑的市场。从全球市场来看,医药定制研发生产外包已经是处于持续快速发展阶段的行业,据IMS预测,到2018年全球医药市场总容量将达到1.3万亿美元。而根据Informa预测,到2017年,中国和印度的CMO市场份额占比也将持续扩大,其中中国的市场份额占比将提升到7.91%,换言之这是一个近千亿美元的巨大市场。MAH制度的施行,使得更多的CMO/CDMO企业一头扎进这一市场之中,竞争博弈。
MAH制度实施到最终,带来的终将会是中国医药生态结构的变化。可以预见的是,对于研发型公司,真正有实力的,凭借MAH所带来的委托生产、委托销售等利好,自然会发展壮大,但一部分靠卖批件为生的研发企业,却将会逐渐失去市场;大型生产企业,要么寻求国内代工厂合作,减少建厂成本,要么便成为国外跨国公司的OEM,从而使生产管理与国际接轨;而至于CMO/CDMO公司,同样恒者恒强,GMP、研发、管理都能做得好的,将迎来巨大的市场,而管理水平落后的,即便有政策利好刺激,也会毫无疑问地死掉。
3
无限挑战
值得注意的是,尽管按照官方口径,MAH制度试点工作开展以来,各试点省市已陆续出台实施方案并取得阶段性成效,但从CFDA的角度来看,“总体工作进度与实际要求还有差距。”为此,2017年8月21日,CFDA再次发文《总局关于推进药品上市许可持有人制度试点工作有关事项的通知》,要求进一步对MAH制度施行的各类问题进行探索。
这并非一个让人意外的情况。事实是,在MAH制度尚未全面推开的当下,持有人的权利义务和法律责任、委托生产中的质量管理体系和生产销售全链条的责任体系、跨区域药品监管机构监管衔接、职责划分以及责任落地等等,都是考验MAH制度中各利益相关方的种种难题。
“首先是国内研发机构的质量管理水平和能力有待提升,其次,目前我国上市后药物警戒管理水平仍处于初级阶段,国家对于这类上市许可持有人的监管将会是很大挑战。”同样以受委托进行工艺研发、临床生产和商业化生产为主业的合全药业副总裁郝玫在接受E药经理人采访时表示。
更为通俗的理解是,被要求对药品进行全生命周期质量负责的药品上市许可持有人,能否承担好这个责任,研发的人能不能管好生产的人,目前存疑。
其实试点方案对上市许可持有人的责任界定得很清楚。就持有人而言,确保注册上市药品风险可控的关键措施是要求其具备药品质量安全责任承担能力,即提供“药物临床试验风险责任承诺书”以及“药品质量安全责任承诺书”等相关文件,以担保协议或者保险合同的形式,确保其质量安全责任承担能力。
然而,单凭一纸承诺是否就可以完成此任务?国内药品生产质量管理技术专家吴军曾走访了大量生产企业,最后发现目前存在的一个最严峻的问题,是企业缺乏对技术的真正理解,还只停留在肤浅的法规政策理解层面。“有的研发企业并不真正了解生产环节,比如说同样都是灌装机,但受托企业的灌装机究竟能不能灌你的产品,很多委托企业并不真的清楚。”
而除此之外,面临最大发展机遇的同时,CMO/CDMO企业同样也正面临着巨大的技术挑战。
“MAH制度更加关注创新,申请者提供的工艺路线不可能简单复制,对受托企业的生产工艺要求较高。如果受托企业仅仅是简单复制,不对工艺做深入研究,那么放大生产失败的概率就会比较大。”杨蕊向E药经理人表示,受托企业如果仅做加工厂是完全不够的,还必须要进行一系列的技术研发、工艺优化等,以确保生产的顺利进行。以凯莱英为例,2016年其仅用于研发方面的投入约为7000万元,占其当年营业收入的6.39%。
“很多企业并没有深入去想。要求看着是在放宽,但我认为是在变相提高。这里面有很多技术的问题,你想当个包工头也不是那么容易的。”吴军笑称,“比如会涉及到生产工艺的调整,你如何去适应不同工厂的要求?比如涉及到供应链的管理,一旦辅料供应换断货了,如何解决更换辅料的问题?再比如如何控制成本问题,要知道企业做研发设计时是不计成本的,但受托企业必须考虑这些问题,要考虑市场的承受能力。”
在解决质量把控、工艺审核、程序厘清等问题过程中,有一个角色不容忽视:保险。
在2016年6月发布的《国务院办公厅关于印发药品上市许可持有人制度试点方案的通知》中,关于保险的要求明确被列入在药品质量安全责任承担能力相关文件之中,要求科研人员或药品研发机构申请成为持有人的,必须承诺在药品上市销售前,向其所在地省级药品监督管理部门提交与担保人签订的担保协议,或者与保险机构签订的保险合同。
一个乐观的情况是,目前有个别地区率先推出配套保险措施,如上海采取“专项风险保险基金+保险”的风险救济制度。由上海张江管委会出资5000万元委托第三方对企业购买的保险进行运营管理,明确对注册在张江高科技园区核心区内的持有人和受托生产企业提供先行风险理赔。此外还有保费补贴,如华领医药、再鼎医药、和记黄埔、海和生物等4家研发企业作为第一批试点企业产生保险总费用共计160余万元,其中符合补贴条件的保险费用共计约140万元,根据此前制定的《操作细则》,拟补贴总额约70万元。
在杨蕊看来,这无疑是十分必要的。“一旦发生药损事件,除了基本的社会保障、社会救济制度的介入,更重要的是有资金赔付。持有人若为药品生产企业,可借助企业资金来化解风险,而对于没有雄厚资金支撑的中小企业、研究机构和科研人员来说,追责起来是没有赔付能力的。”
在这一方面,曾轰动一时的“万络事件”是一起典型案例。美国默沙东所生产的罗非昔布(商品名:万络)此前因严重心血管系统不良反应而撤市并遭遇了大量集体诉讼,最终默沙东宣布愿意以48.5亿美元建立2个以上的基金用于庭外和解万络诉讼案。沈阳药科大学工商管理学院杨悦教授在刚刚结束不久的“药品上市许可持有人制度风险管理”高峰论坛中以此举例,“默沙东为何支付得起高额赔偿?实力是一方面,但同时默沙东购买的商业性产品责任保险,仅2003年保额就高达6.3亿美元。”
“实际上国外在这方面有很多可以借鉴的经验。但就国内而言,对于MAH流程中,哪个阶段适合什么样的保险险种,还需要细致划分,更需要想要投入药品行业的保险公司设计探索。”杨蕊补充道。据了解,目前安达保险、太平洋保险、平安保险等都已陆续推出了相应的责任险。